
Imagine you have a prescription for a life-saving medication. The brand-name version costs $500 a month, but the pharmacy offers a generic alternative for just $15. You take it without hesitation because you know it works exactly the same way. But how do we know that cheap pill is safe? That’s where the Abbreviated New Drug Application, or ANDA, comes in.
An ANDA is not a magic trick; it is a specific legal pathway created by the U.S. government to let companies sell generic drugs without repeating the billions of dollars and years of testing that original drug makers already did. If you are curious about why your medicine has a different color or taste than the brand name, or why some generics appear suddenly while others wait years, understanding the ANDA process clears up the confusion. It is the engine behind affordable healthcare in the United States.
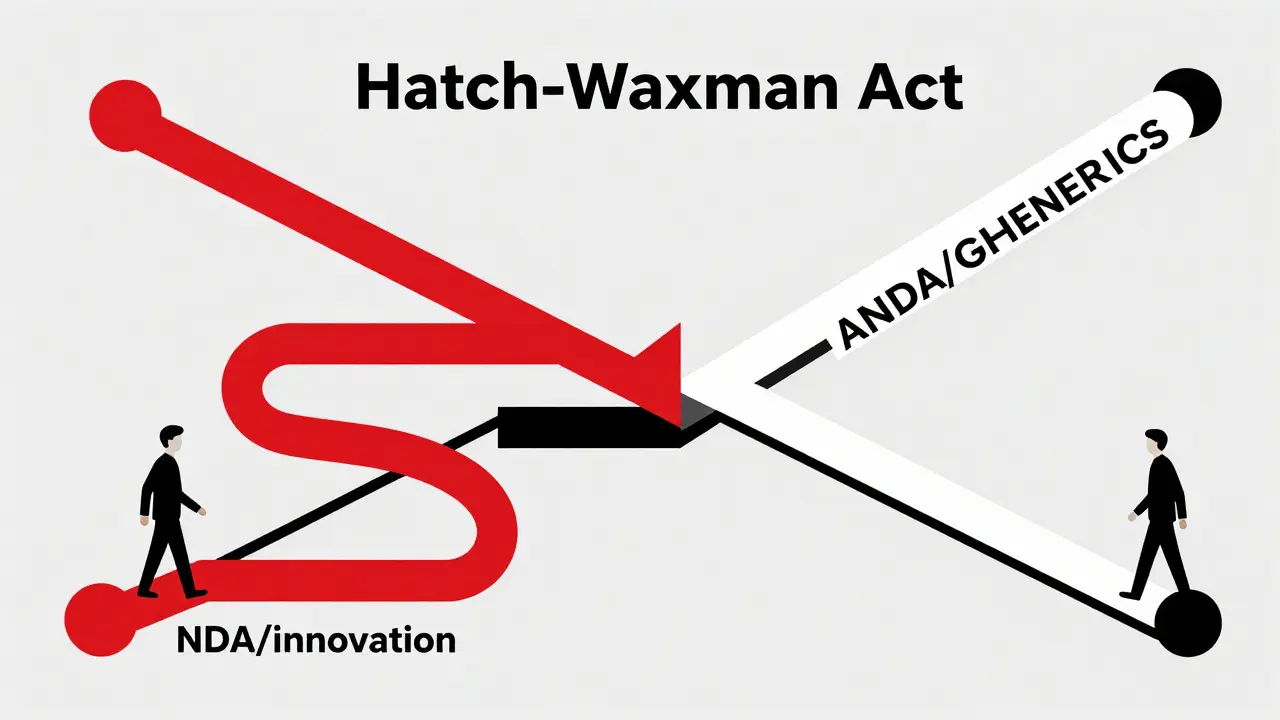
The Origin Story: Why We Need the Hatch-Waxman Act
To understand what an ANDA is, you have to look back at 1984. Before this year, the system was broken. Big pharmaceutical companies held patents on their drugs for so long that generics could never compete. At the same time, small biotech startups struggled to recoup their research costs because they lost patent protection too quickly as they waited for regulatory approval.
The solution was the Drug Price Competition and Patent Term Restoration Act, widely known as the Hatch-Waxman Act. This law created two distinct lanes for drug approval. One lane was for new, innovative drugs (called NDAs), and the other was for generic copies (ANDAs). The goal was simple: balance innovation with access. It allowed generic manufacturers to skip the expensive clinical trials if they could prove their product was equivalent to the brand-name drug. This single piece of legislation saved the U.S. healthcare system trillions of dollars.
How an ANDA Works: The "Abbreviated" Part
The word "abbreviated" does not mean "less rigorous." It means "shorter." When a company wants to launch a new drug from scratch, they file a New Drug Application (NDA). An NDA requires data from animal studies and multiple phases of human clinical trials involving thousands of patients. This takes 10 to 15 years and costs around $2.6 billion.
An ANDA applicant says, "We don’t need to prove this molecule is safe again. The FDA already knows that. We just need to prove our version is the same as yours." Here is what that involves:
- Same Active Ingredient: The generic must contain the exact same chemical compound as the brand-name drug.
- Same Strength and Dosage Form: If the brand is a 10mg tablet, the generic must be a 10mg tablet. You cannot swap a tablet for a capsule unless you go through a much harder process.
- Same Route of Administration: If it is swallowed, it must be swallowed. If it is injected, it must be injected.
- Identical Conditions of Use: It treats the same disease in the same way.
The only things that can differ are inactive ingredients (like dyes or fillers) and packaging. These changes usually do not affect how the drug works, which is why your generic might look slightly different but perform identically.
Bioequivalence: The Core Scientific Test
This is the most critical part of the ANDA process. How does the FDA verify that the generic behaves the same way in your body? They use a test called bioequivalence.
Manufacturers recruit 24 to 36 healthy volunteers. Half get the brand-name drug, and half get the generic. Then, scientists measure how fast and how much of the drug enters the bloodstream. They look at two key numbers:
- AUC (Area Under the Curve): This measures the total amount of drug absorbed over time.
- Cmax (Maximum Concentration): This measures the peak level of the drug in the blood.
For the ANDA to be approved, the generic’s results must fall within a strict range. Specifically, the 90% confidence interval for the ratio of the generic to the brand must be between 80% and 125%. In plain English, the generic must deliver almost the exact same dose to your body at almost the exact same speed. If it is too slow or too fast, the application gets rejected.
The Reference-Listed Drug and the Orange Book
You cannot just pick any drug to copy. You must target a Reference-Listed Drug (RLD). This is the original brand-name drug that the FDA has already approved as safe and effective. To find these drugs, manufacturers consult the FDA Orange Book.
The Orange Book is a public database maintained by the FDA. It lists all approved drug products and includes crucial information about patents and exclusivity periods. If a drug is not in the Orange Book, or if its patents are still active and protected, you generally cannot submit an ANDA for it yet. This book acts as the map for generic developers, showing them which targets are open for competition and which are still locked down by intellectual property laws.
| Feature | ANDA (Generic) | NDA (Brand Name) |
|---|---|---|
| Primary Goal | Demonstrate equivalence to existing drug | Prove safety and efficacy from scratch |
| Clinical Trials | Usually none (except bioequivalence) | Phases 1, 2, and 3 required |
| Development Cost | $1-$5 million | $2.6 billion (average) |
| Time to Market | 3-4 years | 10-15 years |
| FDA Review Timeline | 10 months (under GDUFA) | 10-12 months |
| Data Source | Leverages RLD data | Generates new proprietary data |
Navigating Patents and Exclusivity
Even if a drug is scientifically ready for a generic version, legal barriers often remain. The Hatch-Waxman Act introduced a clever mechanism to handle this. When filing an ANDA, the manufacturer must certify their stance on the patents listed in the Orange Book. There are four types of certifications, known as Paragraph I through IV.
The most dramatic is Paragraph IV certification. This happens when a generic maker claims the brand-name patent is invalid or will not be infringed by their generic product. This triggers a lawsuit from the brand company. During this legal battle, the FDA pauses the approval clock for 30 months. This pause allows the courts to decide who is right.
If the generic company wins, they get a massive reward: 180-day marketing exclusivity. This means they are the sole generic provider for six months before any other competitors can enter. This incentive drives companies to challenge big pharma patents, leading to rapid price drops once the exclusivity period ends.
Challenges in the Modern ANDA Landscape
Not all generics are created equal. Simple pills like ibuprofen are easy to replicate. But what about complex drugs? Think of inhalers for asthma, topical creams for skin conditions, or extended-release capsules that dissolve slowly over 24 hours. These are called complex generics.
Standard bioequivalence tests often fail here. For an inhaler, it doesn’t matter how much drug is in the blood; it matters how much reaches the lungs. Proving this requires specialized studies that cost significantly more and take longer. Recently, the FDA has struggled with backlogs for these complex applications. In 2022, many smaller generic manufacturers reported receiving "Complete Response Letters"-essentially rejection notices due to insufficient data-because their manufacturing controls or bioequivalence data were inadequate.
Additionally, supply chain risks are rising. Many ANDA-approved generics rely on raw materials manufactured in India and China. Geopolitical tensions or local disruptions can threaten the availability of these essential medicines, a concern highlighted by former FDA officials in recent health policy reports.
The Impact on You and the Healthcare System
Why should you care about an ANDA? Because it directly impacts your wallet and your health outcomes. Generic drugs account for approximately 90% of all prescriptions filled in the United States. Yet, they represent only about 23% of total drug spending. This disparity exists because ANDAs allow prices to drop by 80% to 85% compared to brand-name equivalents within the first year of market entry.
This savings is not just theoretical. It keeps insurance premiums lower and ensures that chronic conditions, from diabetes to hypertension, remain treatable for millions of people. Without the ANDA pathway, the U.S. healthcare system would face a financial collapse, and access to basic medications would become a privilege rather than a right.
Can I trust generic drugs approved via ANDA?
Yes. The FDA holds generic drugs to the same high standards as brand-name drugs. They must demonstrate bioequivalence, meaning they work in the body in the same way and provide the same therapeutic effect. The only differences are usually in inactive ingredients like colors or flavors, which do not affect performance.
Why are some generic drugs still expensive?
If a drug has no competing generics, the price stays high. This often happens with "complex generics" that are difficult to manufacture or where only one company has secured the necessary approvals. Once multiple companies enter the market with valid ANDAs, competition drives the price down significantly.
What is the difference between an ANDA and an NDA?
An NDA (New Drug Application) is for brand-new drugs that require full clinical trials to prove safety and efficacy. An ANDA (Abbreviated New Drug Application) is for generic versions of already-approved drugs. The ANDA is "abbreviated" because it relies on the existing safety data of the brand-name drug, requiring only proof of bioequivalence.
What is the FDA Orange Book?
The Orange Book is a publication by the FDA that lists all approved drug products with therapeutic equivalence evaluations. It also contains patent information and exclusivity data for reference-listed drugs. Generic manufacturers use this book to identify which drugs they can legally copy and when patents expire.
How long does it take to get an ANDA approved?
Under the Generic Drug User Fee Amendments (GDUFA), the FDA aims to review standard ANDAs within 10 months. However, the entire development process, including formulation and bioequivalence studies, typically takes 3 to 4 years. Delays can occur if the FDA requests additional data or if patent litigation triggers a 30-month stay.
What is bioequivalence?
Bioequivalence is a scientific comparison that shows the rate and extent to which the active ingredient in a generic drug becomes available at the site of action in the body. For an ANDA to be approved, the generic must show that its bioavailability falls within 80-125% of the brand-name drug's metrics for both AUC and Cmax.




